
https://bcbio.wordpress.com/2013/05/22/scaling-variant-detection-pipelines-for-whole-genome-sequencing-analysis/?utm_source=feedlyBlue Collar Bioinformatics
Scaling variant detection pipelines for whole genome sequencing analysis
with 2 comments
Scaling for whole genome sequencing
Moving from exome to whole genome sequencing introduces a myriad of scaling and informatics challenges. In addition to the biological component of correctly identifying biological variation, it’s equally important to be able to handle the informatics complexities that come with scaling up to whole genomes.
At Harvard School of Public Health, we are processing an increasing number of whole genome samples and the goal of this post is to share experiences scaling the bcbio-nextgen pipeline to handle the associated increase in file sizes and computational requirements. We’ll provide an overview of the pipeline architecture in bcbio-nextgen and detail the four areas we found most useful to overcome processing bottlenecks:
- Support heterogeneous cluster creation to maximize resource usage.
- Increase parallelism by developing flexible methods to split and process by genomic regions.
- Avoid file IO and prefer streaming piped processing pipelines.
- Explore distributed file systems to better handle file IO.
This overview isn’t meant as a prescription, but rather as a description of experiences so far. The work is a collaboration between the HSPH Bioinformatics Core, the research computing team at Harvard FAS and Dell Research. We welcome suggestions and thoughts from others working on these problems.
Pipeline architecture
The bcbio-nextgen pipeline runs in parallel on single multicore machines or distributed on job scheduler managed clusters like LSF, SGE, and TORQUE. The IPython parallel framework manages the set up of parallel engines and handling communication between them. These abstractions allow the same pipeline to scale from a single processor to hundreds of node on a cluster.
The high level diagram of the analysis pipeline shows the major steps in the process. For whole genome samples we start with large 100Gb+ files of reads in FASTQ or BAM format and perform alignment, post-alignment processing, variant calling and variant post processing. These steps involve numerous externally developed software tools with different processing and memory requirements.
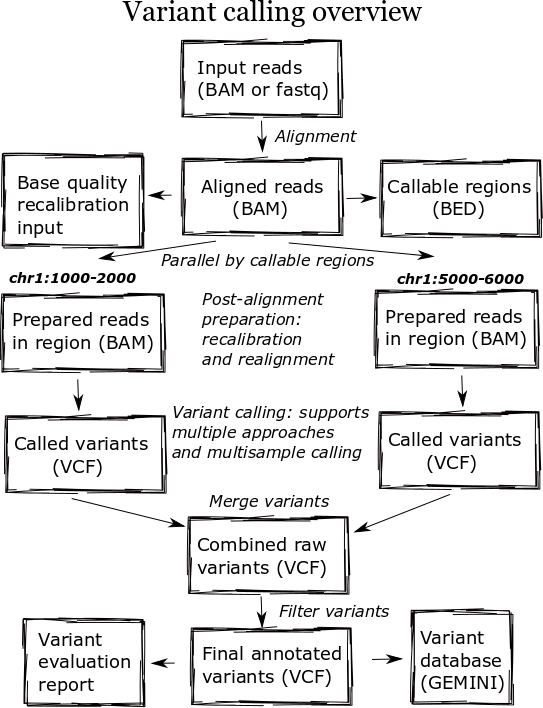
Heterogeneous clusters
A major change in the pipeline was supporting creation of heterogeneous processing environments targeted for specific programs. This moves away from our previous architecture, which attempted to flatten processing and utilize single cores throughout. Due to algorithm restrictions, some software requires the entire set of reads for analysis. For instance,GATK’s base quality recalibrator uses the entire set of aligned reads to accurately calculate inputs for read recalibration. Other software operates more efficiently on entire files: the alignment step scales better by running using multiple cores on a single machine, since the IO penalty for splitting the input file is so severe.
To support this, bcbio-nextgen creates an appropriate type of cluster environment for each step:
- Multicore: Allocates groups of same machine processors, allowing analysis of individual samples with multiple cores. For example, this enables running bwa alignment with 16 cores on multiprocessor machines.
- Full usage of single cores: Maximize usage of single cores for processes that scale beyond the number of samples. For example, we run variant calling in parallel across subsets of the genome.
- Per sample single core usage: Some steps do not currently parallelize beyond the number of samples, so require a single core per sample.
IPython parallel provides the distributed framework for creating these processing setups, working on top of existing schedulers like LSF, SGE and TORQUE. It creates processing engines on distributed cores within the cluster, usingZeroMQ to communicate job information between machines.
Cluster schedulers allow this type of control over core usage, but an additional future step is to include memory and disk IO requirements as part of heterogeneous environment creation. Amazon Web Services allows selection of exact memory, disk and compute resources to match the computational step. Eucalyptus and OpenStack bring this control to local hardware and virtual machines.

Parallelism by genomic regions
While the initial alignment and preparation steps require analysis of a full set of reads due to IO and algorithm restrictions, subsequent steps can run with increased parallelism by splitting across genomic regions. Variant detection algorithms do require processing continuous blocks of reads together, allowing local realignment algorithms to correctly characterize closely spaced SNPs and indels. Previously, we’d split analyses by chromosome but this has the downside of tying analysis times to chromosome 1, the largest chromosome.
The pipeline now identifies chromosome blocks without callable reads. These blocks group by either genomic features like repetitive hard to align sequence or analysis requirements like defined target regions. Using the globally shared callable regions across samples, we fraction the genome into more uniform sections for processing. As a result we can work on smaller chunks of reads during time critical parts of the process: applying base recalibration, de-duplication, realignment and variant calling.

Streaming pipelines
A key bottleneck throughout the pipeline is disk usage. Steps requiring reading and writing large BAM or FASTQ files slow down dramatically once they overburden disk IO, distributed filesystem capabilities or ethernet connectivity between storage nodes. A practical solution to this problem is to avoid intermediate files and use unix pipes to stream results between processes.
We reworked our alignment step specifically to eliminate these issues. The previous attempt took a disk centric approach that allowed scaling out to multiple single cores in a cluster. We split an input FASTQ or BAM file into individual chunks of reads, and then aligned each of these chunks independently. Finally, we merged all the individual BAMs together to produce a final BAM file to pass on to the next step in the process. While nicely generalized, it did not scale when running multiple concurrent whole genomes.
The updated pipeline uses multicore support in samtools and aligners like bwa-mem and novoalign to pipe all steps as a stream: preparation of input reads, alignment, conversion to BAM and coordinate sorting of aligned reads. This results in improved scaling at the cost of only being able to increase single sample throughput to the maximum processors on a machine.
More generally, the entire process creates numerous temporary file intermediates that are a cause of scaling issues. Commonly used best-practice toolkits like Picard and GATK primarily require intermediate files. In contrast, tools in theMarth lab’s gkno pipeline handle streaming input and output making it possible to create alignment post-processing pipelines which minimize temporary file creation. As a general rule, supporting streaming algorithms amenable to piping can ameliorate file load issues associated with scaling up variant calling pipelines. This echos the focus on streaming algorithms Titus Brown advocates for dealing with large metagenomic datasets.
Distributed file systems
While all three of CPU, memory and disk speed limit individual steps during processing, the hardest variable to tweak is disk throughput. CPU and memory limitations have understandable solutions: buy faster CPUs and more memory. Improving disk access is not as easily solved, even with monetary resources, as it’s not clear what combination of disk and distributed filesystem will produce the best results for this type of pipeline.
We’ve experimented with NFS, GlusterFS and Lustre for handling disk access associated with high throughput variant calling. Each requires extensive tweaking and none has been unanimously better for all parts of the process. Much credit is due to John Morrissey and the research computing team at Harvard FAS for help performing incredible GlusterFS and network improvements as we worked through scaling issues, and Glen Otero, Will Cottay and Neil Klosterman at Dell for configuring an environment for NFS and Lustre testing. We can summarize what we’ve learned so far in two points:
- A key variable is the network connectivity between storage nodes. We’ve worked with the pipeline on networks ranging from 1 GigE to InfiniBand connectivity, and increased throughput delays scaling slowdowns.
- Different part of the processes stress different distributed file systems in complex ways. NFS provides the best speed compared to single machine processing until you hit scaling issues, then it slows down dramatically. Lustre and GlusterFS result in a reasonable performance hit for less disk intensive processing, but delay the dramatic slowdowns seen with NFS. However, when these systems reach their limits they hit a slowdown wall as bad or worse than NFS. One especially slow process identified on Gluster is SQLite indexing, although we need to do more investigation to identify specific underlying causes of the slowdown.
Other approaches we’re considering include utilizing high speed local temporary disk, reducing writes to long term distributed storage file systems. This introduces another set of challenges: avoiding stressing or filling up local disk when running multiple processes. We’ve also had good reports about using MooseFS but haven’t yet explored setting up and configuring another distributed file system. I’d love to hear experiences and suggestions from anyone with good or bad experiences using distributed file systems for this type of disk intensive high throughput sequencing analysis.
A final challenge associated with improving disk throughput is designing a pipeline that is not overly engineered to a specific system. We’d like to be able to take advantage of systems with large SSD attached temporary disk or wonderfully configured distributed file systems, while maintaining the ability scale on other systems. This is critical for building a community framework that multiple groups can use and contribute to.
Timing results
Providing detailed timing estimates for large, heterogeneous pipelines is difficult since they will be highly depending on the architecture and input files. Here we’ll present some concrete numbers that provide more insight into the conclusions presented above. These are more useful as a side by side comparison between approaches, rather than hard numbers to predict scaling on your own systems.
In partnership with Dell Solutions Center, we’ve been performing benchmarking of the pipeline on dedicated cluster hardware. The Dell system has 32 16-core machines connected with high speed InfiniBand to distributed NFS and Lustre file systems. We’re incredibly appreciative of Dell’s generosity in configuring, benchmarking and scaling out this system.
As a benchmark, we use 10x coverage whole genome human sequencing data from the Illumina platinum genomesproject. Detailed instructions on setting up and running the analysis are available as part of the bcbio-nextgen example pipeline documentation.
Below are wall-clock timing results, in total hours, for scaling from 1 to 30 samples on both Lustre and NFS fileystems:
| primary | 1 sample | 1 sample | 1 sample | 30 samples | 30 samples | |
|---|---|---|---|---|---|---|
| bottle | 16 cores | 96 cores | 96 cores | 480 cores | 480 cores | |
| neck | Lustre | Lustre | NFS | Lustre | NFS | |
| alignment | cpu/mem | 4.3h | 4.3h | 3.9h | 4.5h | 6.1h |
| align post-process | io | 3.7h | 1.0h | 0.9h | 7.0h | 20.7h |
| variant calling | cpu/mem | 2.9h | 0.5h | 0.5h | 3.0h | 1.8h |
| variant post-process | io | 1.0h | 1.0h | 0.6h | 4.0h | 1.5h |
| total | 11.9h | 6.8h | 5.9h | 18.5h | 30.1h |
Some interesting conclusions:
- Scaling single samples to additional cores (16 to 96) provides a 40% reduction in processing time due to increased parallelism during post-processing and variant calling.
- Lustre provides the best scale out from 1 to 30 samples, with 30 sample concurrent processing taking only 1.5x as along as a single sample.
- NFS provides slightly better performance than Lustre for single sample scaling.
- In contrast, NFS runs into scaling issues at 30 samples, proceeding 5.5 times slower during the IO intensive alignment post-processing step.
This is preliminary work as we continue to optimize code parallelism and work on cluster and distributed file system setup. We welcome feedback and thoughts to improve pipeline throughput and scaling recommendations.