
OK, let me go back to this post. things to remember:
1) You must quality control your fastq files first by fastqc. Trim the adapters and low quality bases by trimmomatic.
2) The fastq files dumped from the SRA GEO repository are usually the raw sequence, you need to quality control them.
3) google, google before you ask anyone else!
Let's start.
inspecting the first several reads of the fastq (this is a ChIP-seq data), I found the quality is coded as phred33
@SRR866627_nutlin_p53.1 HWI-ST571:161:D0YP4ACXX:3:1101:1437:2055 length=51
NCGAAAGACTGCTGGCCGACGTCGAGGTCCCGATTGTCGGCGTCGGCGGCA
+SRR866627_nutlin_p53.1 HWI-ST571:161:D0YP4ACXX:3:1101:1437:2055 length=51
#1:AABDDH<?CF<CFHH@D:??FAEE6?FHDAA=@CF@4@EBA88@;575
@SRR866627_nutlin_p53.2 HWI-ST571:161:D0YP4ACXX:3:1101:1423:2163 length=51
GAAATACTGTCTCTACTAAAAATACAAAAAGTAGCCAGGCGTCGTGGTGCA
+SRR866627_nutlin_p53.2 HWI-ST571:161:D0YP4ACXX:3:1101:1423:2163 length=51
B@CFFFFFHHHHGIJJJJIJJJIJJJJIJJIEGHIJJJJIJIIIHIE=FGC
SSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSS..................................................... ..........................XXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXX...................... ...............................IIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIII...................... .................................JJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJ...................... LLLLLLLLLLLLLLLLLLLLLLLLLLLLLLLLLLLLLLLLLL.................................................... !"#$%&'()*+,-./0123456789:;<=>?@ABCDEFGHIJKLMNOPQRSTUVWXYZ[\]^_`abcdefghijklmnopqrstuvwxyz{|}~ | | | | | | 33 59 64 73 104 126 0........................26...31.......40 -5....0........9.............................40 0........9.............................40 3.....9.............................40 0.2......................26...31.........41 S - Sanger Phred+33, raw reads typically (0, 40) X - Solexa Solexa+64, raw reads typically (-5, 40) I - Illumina 1.3+ Phred+64, raw reads typically (0, 40) J - Illumina 1.5+ Phred+64, raw reads typically (3, 40) with 0=unused, 1=unused, 2=Read Segment Quality Control Indicator (bold) (Note: See discussion above). L - Illumina 1.8+ Phred+33, raw reads typically (0, 41)
then I used fastqc to check the quality:




the quality for each base is very good, but the perbase sequence content does not look good, and the adapters are not trimmed as indicated by the overrepresented sequences (highlighted part is part of the Truseq adapter index1).
Then I used Trimmomatic to trim the adapters and the low quality bases.
$ trimmomatic SE in.fastq in_trimmed.fastq ILLUMINACLIP:Truseq_adaptor.fa:2:30:10 LEADING:3 TRAILING:3 SLIDINGWINDOW:4:15 MINLEN:36
$ cat Truseq_adaptor.fa
>TruSeq_Universal_Adapter
AATGATACGGCGACCACCGAGATCTACACTCTTTCCCTACACGACGCTCTTCCGATCT
>TruSeq_Adapter_Index_1
GATCGGAAGAGCACACGTCTGAACTCCAGTCACATCACGATCTCGTATGCCGTCTTCTGCTTG
>TruSeq_Adapter_Index_2
GATCGGAAGAGCACACGTCTGAACTCCAGTCACCGATGTATCTCGTATGCCGTCTTCTGCTTG
>TruSeq_Adapter_Index_3
GATCGGAAGAGCACACGTCTGAACTCCAGTCACTTAGGCATCTCGTATGCCGTCTTCTGCTTG
>TruSeq_Adapter_Index_4
GATCGGAAGAGCACACGTCTGAACTCCAGTCACTGACCAATCTCGTATGCCGTCTTCTGCTTG
>TruSeq_Adapter_Index_5
GATCGGAAGAGCACACGTCTGAACTCCAGTCACACAGTGATCTCGTATGCCGTCTTCTGCTTG
>TruSeq_Adapter_Index_6
GATCGGAAGAGCACACGTCTGAACTCCAGTCACGCCAATATCTCGTATGCCGTCTTCTGCTTG
>TruSeq_Adapter_Index_7
GATCGGAAGAGCACACGTCTGAACTCCAGTCACCAGATCATCTCGTATGCCGTCTTCTGCTTG
>TruSeq_Adapter_Index_8
GATCGGAAGAGCACACGTCTGAACTCCAGTCACACTTGAATCTCGTATGCCGTCTTCTGCTTG
>TruSeq_Adapter_Index_9
GATCGGAAGAGCACACGTCTGAACTCCAGTCACGATCAGATCTCGTATGCCGTCTTCTGCTTG
>TruSeq_Adapter_Index_10
GATCGGAAGAGCACACGTCTGAACTCCAGTCACTAGCTTATCTCGTATGCCGTCTTCTGCTTG
>TruSeq_Adapter_Index_11
GATCGGAAGAGCACACGTCTGAACTCCAGTCACGGCTACATCTCGTATGCCGTCTTCTGCTTG
>TruSeq_Adapter_Index_12
GATCGGAAGAGCACACGTCTGAACTCCAGTCACCTTGTAATCTCGTATGCCGTCTTCTGCTTG
after that, I checked the with fastqc again:


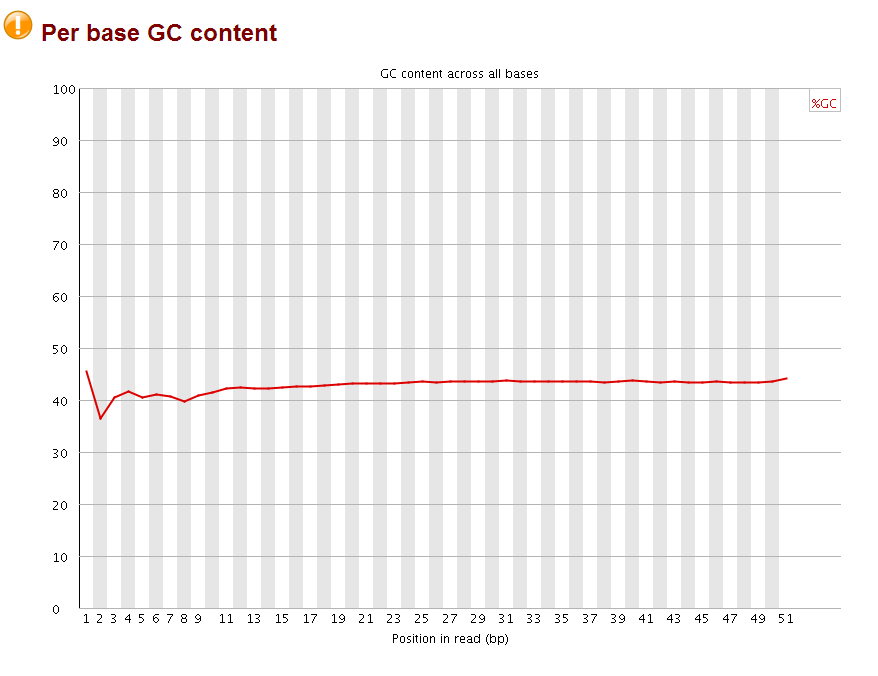
sometime ago, I quality controlled a RNA-seq data, and found found many wired things and asked on biostar.




Take home messages:
1)
"The weird per-base content is extremely normal and is typically referred to as the "random" hexamer effect, since things aren't actually amplified in a uniform manner. Most people will have this and it's nothing to worry about. BTW, don't let anyone talk you into trimming that off, it's the legitimate sequence."
2)
"Duplicates are expected in RNAseq. Firstly, your samples are probably full of rRNAs, even if you performed ribodepletion. Secondly, any highly expressed gene is going to have apparent PCR duplication due solely to it being highly expressed (there is a ceiling on the coverage you can get before running into this, it's pretty high with paired-end reads but not infinite)."
3)Several posts for whether the duplicates need to be removed or not.
https://www.biostars.org/p/55648/
https://www.biostars.org/p/66831/
https://www.biostars.org/p/14283/
Currently, I do not remove them. That's all for today!